-
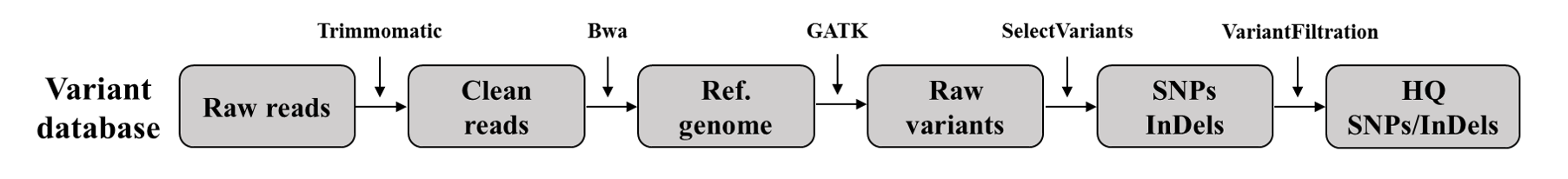
Pipeline for variant calling
Reads filter:We trimmed the adaptor and low-quality sequences in the raw data using Trimmomatic.
Mapping: We mapped the clean reads to the reference genome using BWA.
Duplicated reads trimmer: We removed duplicated reads using Picard.
Calling variants: We performed variant calling using GATK with the HaplotypeCaller module.
HQ variants: We divided the variant database between the SNP and InDel database, and then filtered them into HQ variants using the VariantFiltration module.
-
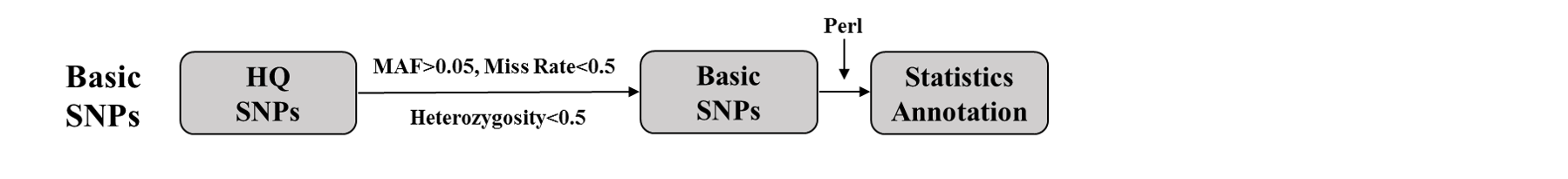
Pipeline for basic SNPs
Basic SNPs: We filtered HQ SNPs with MAF > 0.05, missing rate < 0.5, and heterozygosity < 0.5.
SNP annotation: We counted the MAF, PIC, heterozygosity, and missing rate of each SNP and annotated them using ANNOVAR.
-
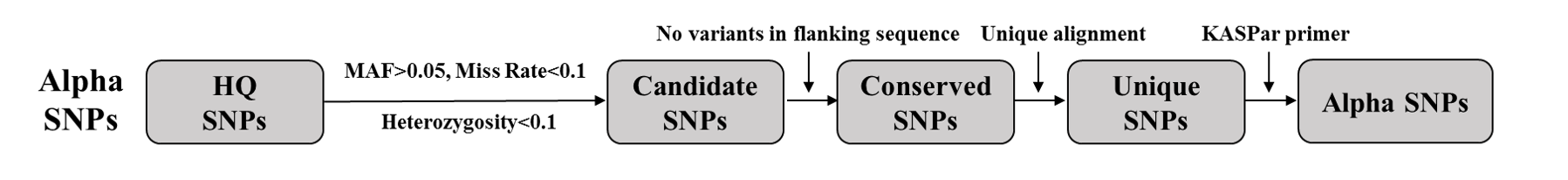
Pipeline for alpha SNPs
Candidate SNPs: We filtered the HQ SNPs into candidate SNPs with MAF > 0.05, missing rate < 0.1, and heterozygosity < 0.1.
Conserved SNPs: We isolated SNPs with no variation from the 30-bp flanking sequence of the candidate SNPs using HQ SNPs and InDels. These SNPs formed the conserved SNP database.
Unique SNPs: We aligned the 30-bp SNP flanking sequence to the reference genome using BLAST (E value = 1e-15). We retained SNPs whose flanking sequences were uniquely aligned to the genome.
Alpha SNPs: We extracted SNPs suitable for the KASPar platform.
-
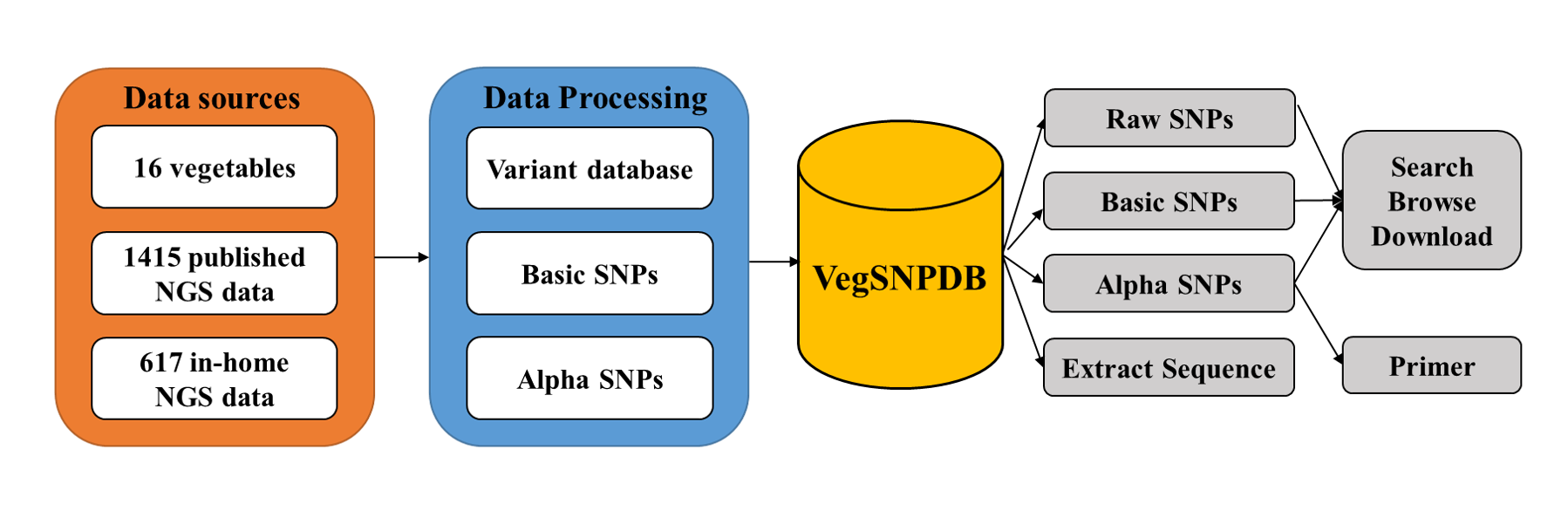
VegSNPDB construction
Data sources: We built VegSNPDB based on published NGS data from 1,415 accessions and in-house NGS data from 617 accessions.
Data processing: Data processing included variant calling, basic SNPs, and alpha SNPs. The detailed pipeline of each module is described above.
VegSNPDB functions:VegSNPDB provides functions for SNP searching, primer design, and sequence extraction.